Introduction to DNA Methylation
DNA methylation constitutes a pivotal epigenetic modification mechanism that involves the enzymatic activity of DNA methyltransferases (DNMTs), which catalyze the addition of methyl groups to specific regions of the DNA molecule, particularly CpG islands. CpG islands are defined as DNA sequences rich in CpG dinucleotides (cytosine-phosphate-guanine sites). While DNA methylation does not alter the underlying nucleotide sequence, it plays a crucial role in the regulation of gene expression.
DNA methylation has broad applications across diverse fields, including:
- Cancer Research: In promoter regions of tumor suppressor genes, hypermethylation can lead to transcriptional silencing, thereby promoting the proliferation, migration, and invasion of tumor cells.
- Neurodevelopment: During embryonic brain development, DNA methylation is involved in the regulation of neural stem cell differentiation and neuronal maturation.
- Neurodegenerative Disorders: In diseases such as Alzheimer's disease, changes in the methylation levels of genes associated with cognitive function have been observed.
- Plant Breeding: Through the modulation of methylation in genes related to stress tolerance, plants can be bred to enhance resilience to environmental stresses, including drought and saline conditions.
CD Genomics currently offers a range of DNA methylation products and services, encompassing several technologies: Whole Genome Bisulfite Sequencing (WGBS), Reduced Representation Bisulfite Sequencing (RRBS), Enzymatic Methyl-seq (EM-seq), and methylation microarrays (850K/935K), ei al,. Each technology exhibits distinct principles and applications, which will be systematically analyzed in this article to assist researchers in selecting the most suitable method based on specific research requirements.
Summary of DNA Methylation Technology Comparisons
| Technology | WGBS | RRBS | EM-seq | 850K/935K Microarrays | Methylation Screening Array (MSA 270K) |
| Detection Principle | Bisulfite conversion coupled with high-throughput sequencing | Dual-enzyme digestion followed by bisulfite conversion and high-throughput sequencing | Enzymatic conversion with high-throughput sequencing | Chip-based analysis using bisulfite-converted DNA | Chip-based analysis using bisulfite-converted DNA |
| Sample Applicability | Applicable to any species with a genome | Restricted to mammalian tissues | Applicable to any species with a genome | Human samples only | Human samples only |
| Required DNA Input | 1–5 μg | 3–5 μg | 200 ng | 3–5 μg | 3–5 μg |
| FFPE Compatibility | Yes | Yes | Yes | Yes | Yes |
| Detection Scope | Genome-wide | Promoter regions; approximately 60% of CpG islands, covering 10–15% of the genome | Genome-wide | Targeted known loci (850K/935K sites), covering approximately 3–4% of the genome | Targeted known loci (270K sites), covering relevant traits and disease phenotypes across a wide range of domains |
Comparison of Technical Principles
WGBS for Genome-Wide DNA Methylation Profiling
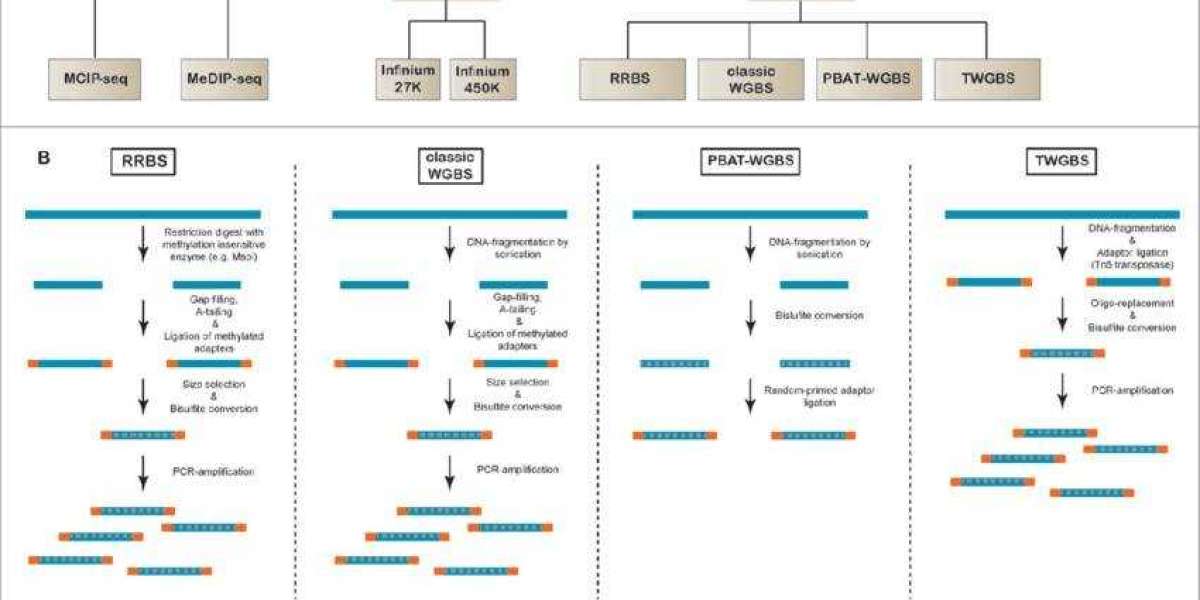
WGBS is a comprehensive technique for detecting DNA methylation across the entire genome. The method relies on the chemical conversion of unmethylated cytosine residues (C) to uracil (U) through bisulfite treatment, while methylated cytosine residues (5-mC) remain unmodified. During subsequent polymerase chain reaction (PCR) amplification, uracil is replaced by thymine (T), allowing differentiation between methylated and unmethylated cytosines upon high-throughput sequencing and alignment to a reference genome. This approach provides a single-base resolution methylation profile across the entire genome.
Advantages
WGBS is among the highest-resolution techniques available for methylation detection, offering an extensive and detailed methylation landscape at a genome-wide scale. The method excels in capturing the breadth of methylation patterns, including novel methylation sites and regions, thereby making it invaluable for exploratory studies seeking to discover new epigenomic markers.
Limitations
The approach requires relatively high amounts of starting DNA (1–5 μg), making it less suitable for low-input samples. WGBS is also associated with considerable technical complexity, high operational costs, and extensive data analysis requirements due to the large volume of sequencing data generated.]
RRBS for Targeted DNA Methylation Analysis
Reduced representation bisulfite sequencing (RRBS) is a targeted DNA methylation sequencing method that selectively enriches DNA fragments containing CpG islands through restriction enzyme digestion. These CpG-rich fragments typically represent methylation-regulated regions, such as gene promoters, which are functionally significant for gene regulation. Following enrichment, fragments undergo bisulfite treatment and sequencing to capture methylation patterns within these targeted regions. This approach reduces genomic complexity while concentrating on methylation sites with known regulatory relevance.
Advantages
RRBS reduces both sequencing costs and data volume by focusing on highly methylated regions of the genome, such as CpG islands and specific methylation contexts (e.g., CHG and CHH sites). This targeted analysis is highly effective for examining methylation changes associated with gene expression regulation. The dual-enzyme digestion strategy (using MspI and ApeKI) improves coverage and accuracy of methylation analysis by enhancing fragment diversity.
Limitations
RRBS is currently optimized for animal samples and requires a relatively high starting amount of DNA (1–5 μg). It does not provide coverage of the entire genome, and thus, certain genomic regions may remain unassessed.
EM-seq for Whole-Genome DNA Methylation Analysis
Enzymatic Methyl-seq is a highly efficient methodology for detecting DNA methylation at single-base resolution across the entire genome. This technique leverages the enzymatic action of Tet methylcytosine dioxygenase 2 (TET2) and T4 bacteriophage beta-glucosyltransferase (T4-BGT) to protect 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) from deamination. Following this protection step, the APOBEC3A enzyme selectively converts unmodified cytosine residues to uracil, while methylated cytosine residues remain unaltered. During polymerase chain reaction (PCR) amplification, uracil is subsequently replaced by thymine, thus enabling differentiation of methylated versus unmethylated cytosine residues through high-throughput sequencing of PCR products. By aligning these sequenced reads with a reference genome, methylation statuses at CpG, CHG, and CHH sites can be precisely determined.
Advantages
EM-seq offers several advantages over traditional bisulfite sequencing (BS-seq) for DNA methylation analysis. This technique requires a lower quantity of starting DNA, with as little as 200 ng, making it suitable for low-input samples. EM-seq also preserves high conversion efficiency without the DNA damage commonly associated with bisulfite treatment, which can cause fragmentation and selective enrichment issues that compromise sequence integrity. By eliminating these artifacts, EM-seq enables cost-effective sequencing while providing high-fidelity methylation data with genome-wide coverage. This approach is particularly advantageous for studies involving plant DNA, where DNA extraction is often challenging, as well as other applications involving low-quantity sample types.
Limitations
As a recently developed technology, EM-seq has limited validation outside human and murine models. Although several plant and non-model organisms have been successfully processed, including Brassica leaves, Myrica fruit, Rehmannia root, and Arabidopsis thaliana, additional optimization and validation are required to expand its applicability across a broader range of species.
DNA Methylation Microarray Technologies: 850K, 935K, and 270K Arrays
DNA methylation microarrays provide a high-throughput approach to assess methylation status at targeted CpG sites by hybridizing bisulfite-treated DNA with designed probes. This technique requires 0.5–1 μg of genomic DNA, which is first subjected to bisulfite conversion to distinguish methylated from unmethylated cytosines. Two types of methylation-specific probes are then hybridized with the converted DNA: one specific to methylated cytosine and the other specific to unmethylated cytosine. Probes hybridize at the 3' CpG position with labeled nucleotides (ddNTPs) and are marked via fluorescent staining. Using the Illumina iScan platform, fluorescence intensity ratios are measured to quantify methylation levels.
The 850K array covers over 850,000 CpG sites, while the enhanced 935K array provides coverage of 935,000 CpG sites. The 270K array, in contrast, includes a subset of 270,000 CpG loci. Given that the human genome contains approximately 28 million CpG sites, with an estimated 60–80% being methylated (equivalent to about 5% of all CpG sites), these arrays provide substantial genome-wide methylation profiling.
 Figure 3 Infinium Methylation Screening Array (image source illumina)
Figure 3 Infinium Methylation Screening Array (image source illumina)
Advantages
Compared to whole-genome methylation sequencing, DNA methylation arrays require lower DNA input (0.5–1 μg), exhibit a shorter analysis cycle, and incur reduced costs. Furthermore, microarray technology is compatible with formalin-fixed, paraffin-embedded (FFPE) samples, making it suitable for large-scale studies, such as those involving hundreds to thousands of samples.
Limitations
Current DNA methylation microarrays are restricted to human samples and are limited to detecting preselected, fixed methylation sites, which may not encompass the entirety of genomic methylation variability.


